Rev. Colomb. Nefrol. 2019;6(2): 172-178, julio-diciembre de 2019. http:www.revistanefrologia.org
Victor Manuel Mora-Bautista1, Tatiana Alexandra Suárez-Pinto2,Gustavo Adolfo Contreras-Garcia3
1Universidad Industrial de Santander; Clínica Materno Infantil San Luis. Bucaramanga, Colombia
2Departamento de pediatría, Universidad Industrial de Santander; Hospital Universitario de Santander.
Bucaramanga, Colombia
3Facultad de Salud, Universidad Industrial de Santander, Santander, Bucaramanga, Colombia
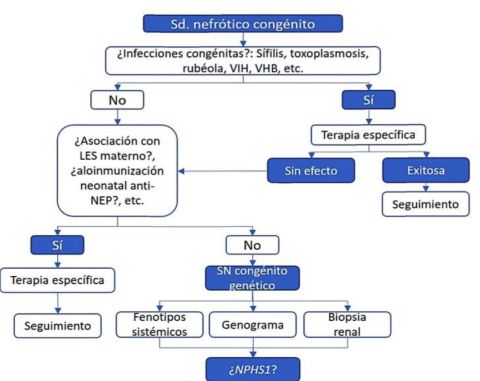
El síndrome nefrótico se define como la unión de proteinuria masiva, hipoalbuminemia e hiperlipidemia, que pueden asociarse a edemas e hipercoagulabilidad. Se origina de una anormalidad de la barrera de filtración glomerular con una fuga masiva de proteína y los efectos secundarios consecuentes. En sus formas primarias, ocurre con una incidencia de 1-3 por cada 100.000 nintildeos menores de 16 antildeos. La forma congénita es una variante poco frecuente del síndrome nefrótico, la cual se presenta en el nacimiento o dentro de los tres primeros meses de vida, y suele ser resistente a la corticoterapia. Se debe evaluar primero la existencia de infecciones congénitas y luego buscar las enfermedades monogénicas más comunes, finalmente se puede recurrir a la secuenciación de nueva generación para buscar mutaciones en los demás genes candidatos. Se presenta el caso de una nintildea con síndrome nefrótico congénito de difícil control, enfatizando en el proceso diagnóstico y el manejo de soporte. Se resalta la importancia de la asesoría genética a la familia en todos los casos.
Palabras clave: síndrome nefrótico congénito, corticoterapia, infecciones congénitas, enfermedades monogénicas, asesoría genética.
A nephrotic syndrome is defined as the association of massive proteinuria, hypoalbuminemia and hyperlipidemia, which may be associated with edema and hypercoagulability. It originates from an abnormality of the glomerular filtration barrier with a massive protein leak and the consequent side effects. In its primary forms, it occurs with an incidence of 1-3 per 100.000 children under 16 years of age. The congenital form is a rare variant of the nephrotic syndrome, which occurs at birth or within the first three months of life and is usually resistant to corticosteroid therapy. Congenital infections and most common related monogenic diseases should be tested. Finally, new generation sequencing must be used to search for mutations in other candidate genes. We present the case of a girl with congenital nephrotic syndrome difficult to control, emphasizing the diagnostic process and support management. The importance of genetic counseling to the family in all cases is highlighted.
Key words: congenital nephrotic syndrome, corticosteroid therapy, congenital infections, monogenic diseases, genetic counseling.
El síndrome nefrótico se define como la asociación de proteinuria mayor a 40 mg/m2/ h (50 mg/kg/d), una albúmina sérica menor de 2,5 g/dL e hiperlipidemia (colesterol total > 200 mg/dL), que pueden asociarse a edemas e hipercoagulabilidad. En su fisiopatología hay una anormalidad de la barrera de filtración glomerular con una fuga masiva de proteína y los efectos secundarios consecuentes1 , 2 Sus formas primarias, ocurren con una incidencia de 1-3 por cada 100.000 nintildeos menores de 16 antildeos3.
La forma congénita es una variante poco frecuente del síndrome nefrótico, la cual se presenta al nacimiento o dentro de los tres primeros meses de vida y suele ser resistente a la corticoterapia 4 , 5 . Establecer un diagnóstico preciso requiere estudios clínicos, bioquímicos, histológicos y genéticos. Las dos causas más frecuentes son el tipo finlandés y la esclerosis mesangial difusa, seguidas de otras enfermedades monogénicas. También se han asociado varias infecciones congénitas, otras causas menos comunes y un grupo de nintildeos donde no se encuentran causas5 , 8.
Se presenta la historia clínica de una nintildea con síndrome nefrótico congénito de difícil control, enfatizando en el proceso diagnóstico del mismo.
Neonato femenina producto de gestación de 33,4 semanas, hija de madre de 25 antildeos, primigestante; con datos prenatales de oligohidramnios, malformación ocular derecha y sospecha de arteria umbilical única, con perfil TORCH negativo. Nació el 10 de julio de 2014 en nuestra institución por cesárea debido a anhidramnios y doppler placentario con redistribución de flujo. Tuvo un peso al nacer de 2.300 g y buena adaptación neonatal. Se confirmó arteria umbilical única en la evaluación física.
Se hospitalizó para estudios por historial antenatal y datos posnatales. El servicio de oftalmología diagnosticó catarata congénita bilateral. Se realizó ecocardiograma que reportó CIA y CIV sin repercusión. Ecografía renal y transfontanelar normales.
Presentó convulsiones al segundo día de vida, yuguladas con fenobarbital. En exámenes, se documentó hipocalcemia, tratada con suplencia transitoria con gluconato de calcio; nitrógeno ureico y creatinina elevados, realizándose seguimiento clínico; y policitemia, por lo que se realizó exanguinotrans-fusión parcial. Ese día, por posible sepsis neonatal temprana le indicaron tratamiento con ampicilina y amikacina. Luego presentó apneas y fue trasladada a la unidad de cuidado intensivo finalizando el mismo día, donde requirió CPAP nasal, mejorando en las 12 horas siguientes.
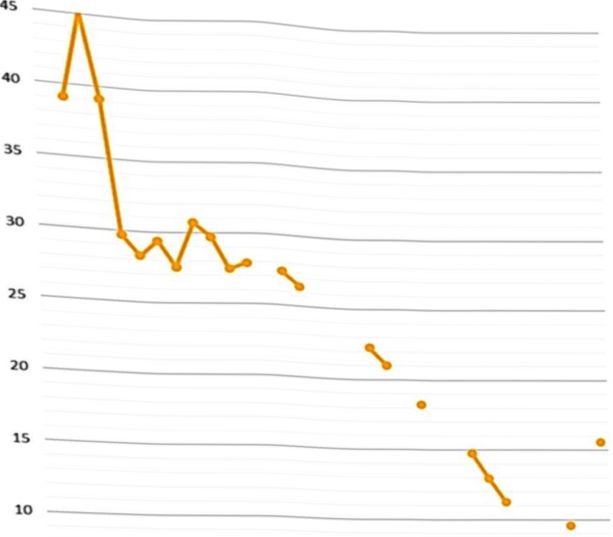
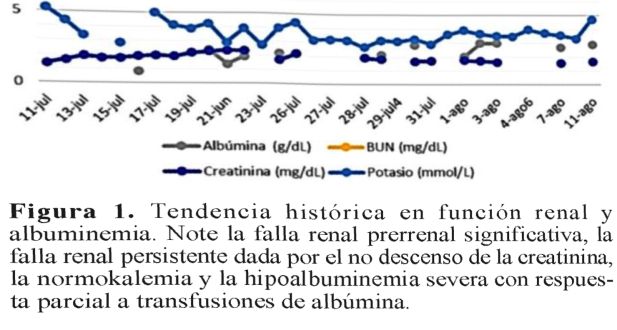
Se diagnosticó insuficiencia renal etapa III, desde el tercer día de vida, con sospecha de displasia renal bilateral. Presentó acidosis metabólica con brecha aniónica elevada, tratada con bicarbonato de sodio. Además, tuvo edema progresivo desde el cuarto día de vida, hasta mostrar anasarca al noveno día. Ante desaparición de las apneas por más de 24 horas, fue trasladada a la unidad neonatal intermedia al sexto día. Se evidenció hipoalbuminemia desde el séptimo día de vida, requiriendo transfusiones de albúmina desde entonces. Ese día se verificó proteinuria en rango nefrótico, falla renal etapa IV (figura 1), hipocalcemia, hipomagnesemia e hiponatremia.
Desde el día 11 de vida, luego de presentar dificultad respiratoria severa por la ascitis (Figura 2), permaneció nuevamente en la unidad de cuidado intensivo. Ese día nefrología pediátrica diagnosticó síndrome nefrótico congénito y solicitó serología para perfil TORCH, perfil tiroideo y laboratorios en búsqueda de autoinmunidad (ANAs, ANCAs, complemento), con resultados normales. Requirió ventilación mecánica desde el día 13 de vida, asociándose nuevo esquema antibiótico (piperacilina-tazobactam) ante el deterioro clínico y oligoanuria. Fue tratada desde entonces con albúmina, antilnpertensivos e infusión de furosemida.
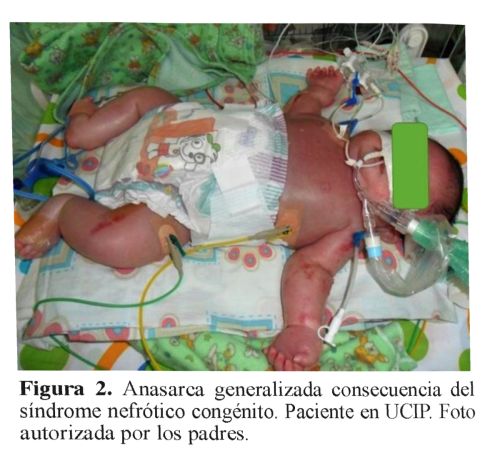
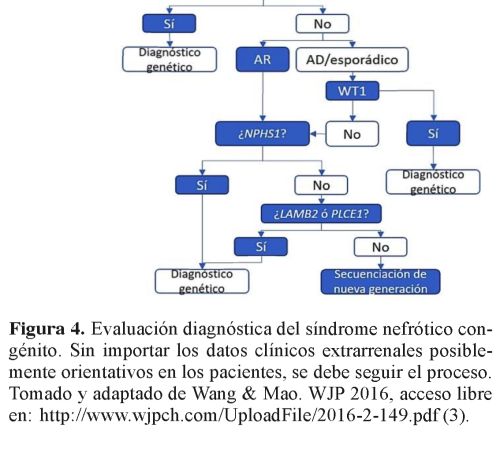
Al día 18 de vida, genética clínica consideró probable síndrome de Pearson, por cursar con síndrome nefrótico congénito, probable microcoria y catarata congénita bilateral. Se solicitó cariotipo y análisis de secuenciación del gen LAMB2, los cuales no fueron realizados por problemas administrativos de su aseguradora. No se logró identificar un patrón de herencia específico a nivel familiar (figura 3).
Al día 20 de vida, nefrología descartó la posibilidad de biopsia renal por su mala condición clínica. En días posteriores su condición clínica permaneció estacionaria. Al día 32 de vida, gradualmente entró en colapso circulatorio hasta fallecer al día siguiente.
Al abordar el síndrome nefrótico congénito, es vital apoyarse en las características semiológicas y comorbilidades para orientar un diagnóstico etiológico clínico; se conoce que las patologías subyacentes comparten varias características, como la poca respuesta al tratamiento y la falla renal, salvo las formas asociadas a infecciones congénitas5
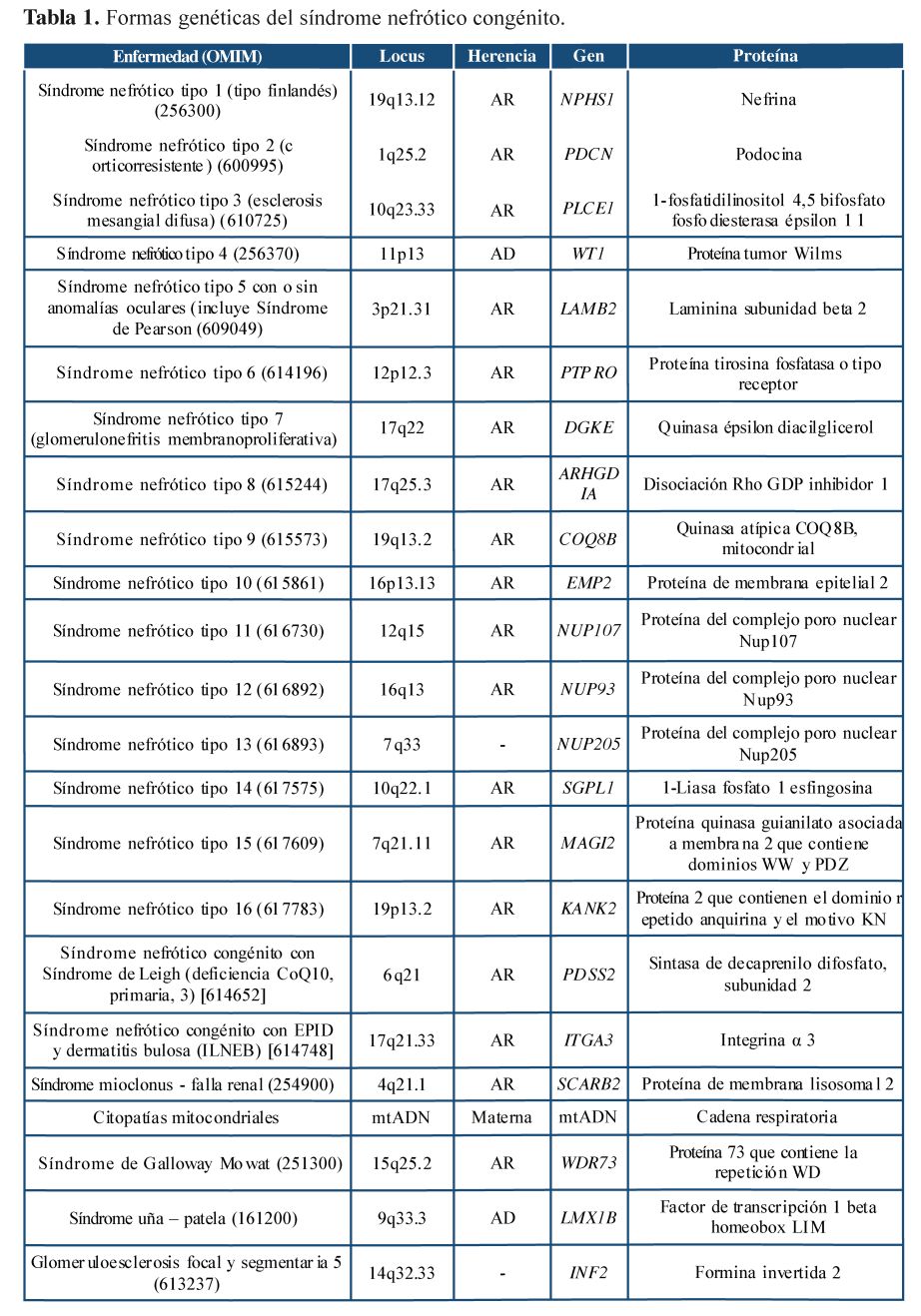
Entre las causas primarias, el tipo finlandés es una enfermedad autosómica recesiva, donde el síndrome nefrótico se presenta en el nacimiento, es severo y no responde al tratamiento, progresando rápidamente a una falla renal. La esclerosis mesangial difusa es la segunda causa de síndrome nefrótico congénito; puede ser una forma aislada o asociarse al síndrome de Denys- Drash, cursando con trastornos de la diferenciación sexual y tumor de Wilms; también es resistente al tratamiento y progresa frecuentemente a falla renal en la infancia. Otras causas incluyen el síndrome de Pearson que se asocia a problemas oculares, el síndrome de Galloway que se relaciona con microcefalia y hernia hiatal, y las mutaciones de podocinas (tabla 1). No se encontraron asociaciones reportadas con arteria umbilical única6 , 7.
Desde 1998 se ha avanzado en la identificación de los genes asociados3. Aunque estos suelen ser monogénicos, se ha reportado un caso con compromiso en más de un gen asociado9. Actualmente se cuenta con técnicas de diagnóstico molecular avanzadas que permiten evaluar varios genes al mismo tiempo como es el uso de la secuenciación de nueva generación o estudio de exorna dirigido 10; el inconveniente continúa siendo el alto costo de los estudios.
Dentro de las causas secundarias se incluyen la sífilis congénita (inicio entre 2o- 3er mes, causa glomerulonefritis membranosa y revierte con el tratamiento), la toxoplasmosis (inicio entre ler-3er mes, proteinuria masiva y revierte con el tratamiento), el citomegalovims (inicio hacia 2° mes, causa esclerosis mesangial difusa y responde a ganciclovir dentro de los primeros 14 días), el virus de Epstein Barr, la rubéola (poco frecuente), la hepatitis B (rara, ocurre a cualquier edad), la malaria, el virus de la inmunodefíciencia humana (suelen ser nintildeos mayores de 1 antildeo), el lupus congénito, trombosis de la vena renal y la aloinmunización materna a la endopepüdasa neutral de los podocitos2 ,3 , 5 , 11.
A nivel imaginológico, antes del nacimiento, es de vital importancia el historial familiar, una placenta engrosada, retraso de crecimiento y el hallazgo de hiperecogenicidad renal. Luego del nacimiento, hay tres patrones que pueden ser oricntativos: áreas hipereco-génicas córtico- medulares con un patrón parcheado y una diferenciación córtico - medular parcial (esclerosis mesangial difusa - patrón glomerular); rintildeones aumentados de tamantildeo, con una corteza hiperecóica y unas pirámides renal que se van encogiendo gradualmente (tipo finlandés-patrón tubular); y unos rintildeones hiperecóicos, agrandados pero con una diferenciación córtico- medular preservada, con una progresión hacia la normalidad (aloinmunización materna). En los otros tipos la ecografía es anormal, pero aún no se han establecido patrones. El hallazgo de un tumor renal sugiere el síndrome de Denys - Drash o WAGR2.
La realización de biopsia renal es controvertida, porque los hallazgos se pueden superponer y el compromiso puede ser diferente por áreas8. Proporciona más información pronostica que diagnóstica. Actualmente se prefiere pasar de una vez al estudio genético. Se presenta un algoritmo diagnóstico en forma ilustrativa (figura 4).
En cuanto al manejo, dado que la mayoría de los casos son resistentes a esteroides, la meta es controlar el edema y la uremia, junto con el manejo de las complicaciones (trombosis, infecciones, raquitismo, hipotiroidismo, anemia, etc.)2 , 5, 12.
Se han usado inhibidores de la enzima convertidora de angiotensina y la indometacina para disminuir la proteinuria, con respuesta parcial y variable. Se ha utilizado también ciclosporina A o tacrolimus, lográndose remisiones parciales, aunque a expensas de sus efectos secundarios4 , 8 , 13 , 14. Otra terapia son las infusiones frecuentes de inmunoglobulina G humana, con buenos resultados en casos de esclerosis mesangial difusa15.
Suele recomendarse la nefrectomía bilateral temprana para controlar la proteinuria; aunque se opta por la nefrectomía unilateral en ambientes de pocos recursos, dado que evita el requerimiento dialítico16.
El trasplante renal es la única posibilidad de curación. Se debe realizar cuándo se tiene más de 10 kg de masa corporal, sin embargo no es accesible 2 , 5 , 16 . En los casos del tipo finlandés, puede aparecer síndrome nefrótico en el riñón trasplantado por circulación de anticuerpos antineurina. El uso de micofenolato, ciclofosfamida oral, dosis altas de metilprednisolona o plasmaféresis (con o sin rituximab) evitan el rechazo 14.
En vista de que no existe un tratamiento curativo para este grupo de patologías a la fecha, la asesoría genética es de vital importancia al establecer el mecanismo de herencia y orientar a las familias sobre el riesgo de recurrencia.
Se declara que no se recibió ningún tipo de remuneración por el desarrollo del presente manuscrito y que no se recibe ninguna contraprestación material de la institución mencionada.
El desarrollo de la presente investigación se realizó con recursos propios de los autores.
Víctor Manuel Mora-Bautista: elección del caso, obtención del consentimiento informado, revisión del tema, redacción y revisión del texto.
Tatiana Alexandra Suárez-Pinto y Gustavo Adolfo Contreras-García: elección del caso, revisión del tema, redacción y revisión del texto.
Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Los autores declaran que en este artículo no aparecen datos de pacientes
1. Román-Ortiz E. Síndrome nefrótico pediátrico. Protoc diagn ter pediatr. 2014;1:285-301.
2. Avni EF, Vandenhoute K, Devriendt A, Ismaili K, Hackx M, Janssen F, et al. Update on congenital nephrotic syndromes and the contribution of US. Pediatr Radiol. 2011;41(1):76-81. https://doi.org/10.1007/s00247-010-1793-5
3. Wang J-J, Mao J-H. The etiology of congenital nephrotic syndrome: current status and challenges. World J Pediatr. 2016;12(2):149-58. https://doi.org/10.1007/sl2519-016-0009-y
4. Mulic B, Milosevski-Lomic G, Paripovic D, Kruscic D, Mulic M, Peco-Antic A. Congenital nephrotic syndrome may respond to cyclosporine a - a case report and review of literature. Srp Arh Celok Lek. 2017;145(7-8):407-10. https://doi.org/10.2298/ SARH160907070M
5. Oliveros Andrade OA, Endo J. Síndrome nefrótico congénito. PRECOP - CCAP. 2018; 17(3): 11-24.
6. Niaudet P. Congenital and infantle nephrotic syndrome. Curr Paediatr. 2006;16(4):264-8.
7. Niaudet P. Congenital nephrotic syndrome. In: Elzouki AY, Harfi HA, Nazer HM, Stapleton FB, Oh W, Whitley RJ, editors. Textbook of Clinical Pediatrics. Berlin, Heidelberg: Springer Berlin Heidelberg; 2012. p. 2793-8.
Disponible en: http://link.springer.com/10.1007/978-3-642-02202-9
8. Fanni C, Loddo C, Faa G, Ottonello G, Puddu M, Fanos V. Congenital nephrotic syndrome. J Pediatr Neonatal Individ Med. 2014;3(2):l-8.
9. Cobos-Carrascosa E, Campos-Aguilera A, Daza-Torres A. Multigene involvement in congenital nephrotic syndrome. Nefrologia. 2014;34(2):268-70. https://doi.org/10.3265/Nefrologia.pre2014
10. GroopmanEE, Rasouly HM, Gharavi AG. Genomic medicine for kidney disease. Nat Rev Nephrol. 2018;14(2):83-104. https://doi.org/l 0.103 8/nrneph.2017.167
11. Soares SF da S, Donatti TL, Souto FJD. Serological Markers of Viral, Syphilitic and Toxoplasmic Infection in Children and Teenagers With Nephrotic Syndrome: Case Series From Mato Grosso State, Brazil. Rev Inst Med Trop Sao Paulo. 2014;56(6):499-504. http://dx.doi.org/10.1590/S0036-46652014000600008
12. Lau KK, Chan HH, Massicotte P, Chan AK. Thrombotic complications of neonates and children with congenital nephrotic syndrome. Curr Pediatr Rev. 2014;10(3):169-76.
13. Büscher AK, Kranz B, Büscher R, Hildebrandt F, Dworniczak B, Pennekamp P, et al. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2010;5(ll):2075-84. http://dx.doi.org/10.2215/CJN.01190210
14. Gulati S, Uttam R. Congenital Nephrotic Syndrome: A Case Report and Review of Fiterature. JIMSA. 2015;28(l):35-6.
15. Gentner JE, Wood EG, Vogler CA, Beck AM, Knutsen AP. Remission of congenital nephrotic syndrome from diffuse mesangial sclerosis with IVIG therapy. J Allergy Clin Immunol. 2015;121(2):83. https://doi.org/10.1016/jjaci.2007.12.331
16. Moodley R, Naicker E, Bhimma R. Congenital nephrotic syndrome: A diagnostic andmanagement dilemma. South African J Child Heal. https://doi.org/10.7196/SAJCH.2015.v9i4.903