http://doi.org/10.22265/acnef.4.1.234
Reporte de caso
Rev. Colomb. Nefrol. 2017;4(1): 99 - 111
Compromiso renal en un paciente con granulomatosis de Wegener: caso clínico y revisión de la literatura
Renal compromise in a patient with Wegener’s granulomatosis: clinical case and literature review
José Luis Fabris1,*, Gilberto Jaramillo1, Rodolfo Torres2, Carlos Rosselli2, Carlos Olivares2, Orlando Olivares2
1Residente de Medicina Interna, Hospital Universitario de San José, Fundación Universitaria de Ciencias de la Salud, Bogotá, Colombia
2Miembro activo del Servicio de Nefrología, Hospital Universitario de San José, Bogotá, Colombia
Referenciar este artículo: Fabris JL, Jaramillo G, Torres R, Rosselli C, Olivares C, Olivares O. Compromiso renal en un paciente con granulomatosis de Wegener: caso clínico y revisión de la literatura. Rev. Colomb. Nefrol. 2017;4(1): 99 - 111.
Correspondencia: *José Luis Fabris Vargas, cjlfabris@fucsalud.edu.co
Recibido: 29-11-16
Aceptado: 25-01-17
Publicado en línea: 20-02-17
Resumen
El presente manuscrito se hace con el fin de reportar un caso del tema en mención y realizar una revisión de la literatura disponible más reciente con respecto al diagnóstico y tratamiento.
Antecedentes
La granulomatosis de Wegener (GW) se caracteriza por ser una vasculitis de pequeños vasos que afecta generalmente la vía respiratoria alta, pulmones y riñones. La afección renal puede presentarse entre el 40 % y el 100 % de los casos. La enfermedad varía en su presentación clínica, desde formas asintomáticas hasta glomerulonefritis rápidamente progresiva, llevando a la insuficiencia renal terminal en pocos días.
Metodología
Se realiza la presentación del caso de una paciente diagnosticada con granulomatosis de Wegener (GW) en el Hospital San José (Bogotá, Colombia), y se muestra una revisión de la literatura enfocada en el compromiso renal de la patología. La revisión bibliográfica se realizó en las bases de datos: PubMEed, LILAC, SciELO y Cochrane, seleccionando para revisión artículos en inglés y español, sin restricción de fecha, pero asegurándose de que el 70 % de las referencias seleccionadas se hubieran publicado después del año 2010.
Conclusiones
El compromiso renal en pacientes con GW es frecuente y puede llegar a ser letal. Una proporción de pacientes considerable puede llegar a requerir terapia de reemplazo renal, con potencial recuperación de la función renal. No están claros los factores que se asocian al retomo de la función renal normal, pero sí se ha descrito que se asocia a tratamiento temprano con altas dosis de inmunomoduladores. El rituximab esun medicamento con uso frecuente en estos pacientes, que ha demostrado efectividad y menores efectos secundarios que los tratamientos convencionales, constituyendo una opción válida para el manejo de pacientes con GW y compromiso renal asociado.
Palabras claves: compromiso renal, eventos adversos, granulomatosis de Wegener (WG), terapia de reemplazo renal.
Absctrat
Wegener’s granulomatosis (WG) is characterized by small vessel vasculitis that affects the upper respiratory tract, lungs and kidneys. Renal involvement may occur between 40 and 100% of cases. The disease varies from asymptomatic to fulminant rapidly progressive glomerulonephritis forms, leading to terminal renal failure in days.
Methodology
We present a case of a patient diagnosed with WG in the Hospital de San José in Bogotá, Colombia, and a literature review focused on renal involvement. Literature search was conducted in the following databases: PubMEed, LILAC, SciELO and Cochrane. Articles in english and Spanish were selected and no restriction date was made, but at least 70% of selected references were published after 2010.
Conclusions
Renal involvement in patients with WG is common and can be lethal. A high percent may even require renal replacement therapy, although patients can return to normal renal function. Factors associated with return to normal renal function are unclear, but it depends on early therapy and high doses of immunomodulators. Rituximab has shown greater effectiveness and fewer side effects than conventionaltreatments, so it’s a valid therapeutic option for the management of patients with WG and renal involvement.
Key words: Renal impairment, Adverse events, Wegener’s granulomatosis, Renal replacement therapy.
Introducción
Las vasculitis sistémicas asociadas a los anticuerpos anticitoplasma de neutrófilos (ANCA), incluyen el síndrome de Churg-Strauss,la granulomatosis de Wegener (GW), la poliangeítis microscópica (PAM), y su forma limitada renal: la glomerulonefritis necrosante idiopática1. La GW se caracteriza por ser una vasculitis necrotizante granulomatosa de pequeños vasos que afecta generalmente a las vías respiratorias altas, los pulmones y riñones.
La enfermedad fue descrita por primera vez en la literatura médica a finales del siglo XIX en un caso clínico. Luego, en la década del 30 del siglo pasado, se nombró por Friedrich Wegener, quien describió la tríada clínica consistente en compromiso pulmonar, renal y de vías respiratorias altas. Desde 2011,la GW se conoce como granulomatosis con poliangeítis (GPA)2. La enfermedad puede presentarse en una forma localizada, que generalmente afecta al tracto respiratorio superior, o sistémica, la cual es más severa y agresiva. Las manifestaciones clínicas dependen de la afectación de los vasos sanguíneos comprometidos y, además de la tríada clásica, también pueden afectar el sistema nervioso centraly periférico, la piel, el tracto gastrointestinal y el sistema musculoesquelético.
La incidencia de la enfermedad se sitúa entre los 2 y los 12 casos por millón de habitantes, con una frecuencia igual en hombres y mujeres. Es muy poco frecuente en la infancia y los adultos jóvenes. El pico de incidencia se reporta en la séptima década de vida, entre los 65 y 70 años de vida, con una prevalencia que está entre los 24 y 157 casos por millón de habitantes2,3. La tasa de incidencia de mortalidad es de 43,5 (IC del 95 %: 35,4 a 52,9)por 1000 personas/año. En el primer año, luego del diagnóstico, la tasa es de 97,4 (IC del 96 %: 69,6 a 132,6) por 1000 personas/año; no obstante, a medida que pasan los años la mortalidad disminuye4.Los pacientes con compromiso renal tienen un mayor riesgo de mortalidad, en especial aquellos con requerimiento de terapia dialítica, donde la mortalidad se incrementa con el paso de los años, siendo las principales causas de ésta los eventos cardiovasculares y las infecciones5-6.
A continuación se realiza la presentación del caso de un paciente con GW atendido en el Hospital San José (Bogotá, Colombia), y una revisión de la literatura enfocada en el compromiso renal de la patología.
Presentación del caso
Se trata de una paciente de 76 años de edad con antecedente de hipertensión arterial crónica, hipotiroidismo, insuficiencia venosa crónica y glaucoma, quien consulta al servicio de urgencias del Hospital San José por cuadro clínico de 1 mes de evolución, consistente en otorrea intermitente fétida bilateral —asociado a rinorrea—, tos con expectoración blanquecina, hiporexia y fiebre. La paciente es valorada por el servicio de otorrinolaringología, quien en la otoscopía bilateral evidencia detritus no fétidos en los conductos auditivos. En la rinoscopia anterior se observan costras nasales, por lo que realizan diagnóstico de otitis media crónica activa y rinosinusitis aguda. Debido a ello los médicos ordenan su hospitalización e iniciar el manejo con ampicilina sulbactam intravenosa y con dexametasona-ciprofloxacina (Fixamicin® Dexacipro) en gotas óticas.
La paciente presenta dificultad respiratoria y taquicardia, con estudio de laboratorios que reportan leucocitosis, anemia, elevación de azoados (creatinina 2,7 mg/dl, BUN 36 mg/dl) y radiografía de tórax con opacidades alveolares que comprometen las regiones parahiliares y básales de ambos pulmones por proceso consolidativo multilobar y derrames pleurales bilaterales libres en escasa cantidad. Se solicita valoración por medicina interna, en donde se evalúa y se plantea diagnóstico de neumonía adquirida en la comunidad severa, con CURB-654, compromiso multilobar y lesión renal aguda AKIN II, por lo que se escalona terapia antimicrobiana a piperacilina/tazobactam, vancomicina; asimismo, al manejo se adiciona oseltamivir, tras sospecha de neumonía viral por H1N1.
Al segundo día de hospitalización la paciente presenta deterioro de cuadro, con falla ventilatoria aguda tipo 1. Se procede a intubación orotraqueal, inicio de ventilación mecánica, soporte vasopresor y traslado a unidad de cuidados intensivos. Se solicita TAC de tórax con hallazgo de áreas con aumento en la densidad del parénquima pulmonar con patrón de vidrio esmerilado y consolidaciones multilobares bilaterales de mayor profusión en lóbulos superiores (especialmente en el lóbulo superior izquierdo) y líquido pleural bilateral libre. Se ordena fibrobroncoscopía por servicio de neumología, con reporte de lavado broncoalveolar: macrófagos: 67%; linfocitos: 0 %; polimorfonuclear: 23 %; Giemsa: negativo para Pneumocystis jirovecii; tinción de Ziehl-Neelsen: negativos para BAAR; Grocott: estructuras con morfología de levaduras y pseudohifas de cándida. Los extendidos en B muestran sobre un fondo limpio presencia de células ciliadas, macrófagos alveolares y ocasionales polimorfonucleares. De otro lado, como no hay células tumorales en este material, el servicio de neumología considera continuar pauta antibiótica y adiciona al tratamiento manejo antifúngico con fluconazol.
El servicio médico evidencia deterioro de la función renal (creatinina 2,9 mg/dl, BUN 51 mg/dl), por lo que solicita interconsulta a nefrología tras la sospecha de necrosis tubular aguda. Se considera que en el contexto de un paciente con antecedentes de infección del tracto respiratorio superior, anemia y alteración de la función renal, debe considerarsela vasculitis de pequeño vaso como primera posibilidad (granulomatosis de Wegener vs poliangeítis microscópica). Es por ello que se solicitan uroanálisis con reporte de: densidad: 1015; pH: 5, nitritosnegativos; proteínas: 150; glucosa normal; células epiteliales: 0-2xc; bacterias escasas; leucocitos: 0-2xc; hematíes: 20-30xc; moco +; cristales de uratos amorfos ++; en el Gram de orina no se observan bacterias; proteínas en orina 24 horas, con reporte de 0,7 gr/24h; ecografía renal y de vías urinarias,con riñones de localización y tamaño usuales, con adecuado grosor cortical y buena diferenciación corticomedular, sin evidencia de lesiones focales intraparenquimatosas; hay aumento difuso en la ecogenicidad del parénquima renal en forma bilateral.
También se ordena estudio inmunológico con resultado de anticuerpos c-ANCA positivo, con título: 1:160 y Coombs directo 2+. Los estudios de Complemento C3, Complemento C4, anti La/SSB, antiRNP, anti Ro/SSA, anti SLC-70 ac, anti Sm (antiSmith), Creatinquinasa MB (CK-MB) y p-ANCA(anticuerpos anticitoplasma de neutrófilos con patrón de fluorescencia perinuclear) presentan resultado dentro de rango normal.
Con el reporte de c-ANCA positivo, además de la clínica referida por la paciente y el rápido deterioro respiratorio, renal y hemodinámico, se confirma el diagnóstico de vasculitis de pequeño vasotipo GW, por lo que se inician bolos de metilprednisolona por 3 días, en el día 14 de la hospitalización. Asimismo se ordena una biopsia renal con estudios de inmunofluorescencia para caracterización de compromiso renal, dada proteinuria y enfermedad renal parenquimatosa difusa.
Se interconsulta al servicio de hematología por sospecha de anemia hemolítica por prueba de Coombs directo positivo y el reporte de frotis de sangre periférica, con glóbulos rojos normocíticos normocrómicos, policromatofilia: 1+, esquistoci-tos: 1+, anisocitosis: 3+. La especialidad considera que la anemia normocítica normocrómica no es deorigen autoinmune, pero dada la progresión agresiva de la clínica se deben descartar procesos neoplásicos hematolinfoides, por lo que solicita estudios de médula ósea y electroforesis de proteínas, con reporte de ausencia de pico monoclonal en electroforesis de proteínas. Con estos resultados el servicio de hematología descarta la patología neoplásica como causa de la anemia.
Se recibe reporte de biopsia renal: parénquima renal con siete glomérulos: cuatro de ellos con esclerosis global y los tres viables con proliferación extracapilar en fase fibrocelular, con proliferación endocapilar y atrapamiento de leucocitos; no hay necrosis ni cariorrexis. Los túbulos presentan atrofia del 20 %, asociada a fibrosis en el mismo porcentaje, con infiltrado inflamatorio mononuclear difuso. En los vasos no hay trombosis, vasculitis ni granulomas. La inmunofluorescencia de 14 glomérulos, 2 esclerosados IgG línea basal Fib, Clq, c3 negativo, acúmulos focales en glomérulos esclerosados IgM, IgA negativo, kappa lineal en un glomérulo, lambda lineal en un glomérulo, albúmina negativo. Los hallazgos muestran una glomerulonefritis proliferativa extracapilar pauciinmune con cambios de cronicidad.
Luego de finalizados los pulsos con esteroide se continúa el manejo con prednisolona (dosis de30 mg/día/vo) y se adiciona manejo citostático con rituximab (dosis de 375 mg/mt2/semana) durante 4 semanas. A dos días de iniciada la terapia oral con prednisolona, la paciente presenta alteración del estado de conciencia asociada a la elevación de azoados, considerándose que cursa con encefalopatía urémica (BUN 118 mg/dl, creatinina 3,4mg/dl). De allí que se indique el inicio de terapia de reemplazo renal, en modalidad de hemodiálisis convencional, con una duración entre 3 y 4 horas diarias. El procedimiento se realizó diariamente,logrando mejorar el estado de conciencia y los signos de sobrecarga hídrica. La paciente se logra extubar al cuarto día de terapia de reemplazo renal; su evolución es satisfactoria y se da egreso de UCI a salas de hospitalización general, donde se continúa terapia dialítica de forma interdiaria, terapia inmunosupresora con prednisolona y dosis programadas de rituximab.
El estudio de patología confirma el diagnóstico: vasculitis de pequeño vaso tipo Granulomatosis de Wegener. Se da salida por estabilización de cuadro y se continúa con el tratamiento ambulatorio de terapia de reemplazo renal más prednisolona (dosisde 30 mg/día vía oral).
Revisión de la literatura y discusión
Etiopatogenia
La GW generalmente inicia como una enfermedad granulomatosa de las vías respiratorias que progresa a una vasculitis sistémica, lo que sugiere una respuesta inmune mediada por células aberrantes a los antígenos (exógenos o endógenos), resultando en la formación de granulomas.
La etiopatogenia autoinmune es compleja, dado que implica la generación de ANCA contra la pro-teinasa 3 (PR3) en aproximadamente el 80 % delos pacientes y contra la mieloperoxidasa (MPO) en aproximadamente el 10 %. Los anticuerpos contra el lisosoma asociado a membrana de proteína-2(LAMP-2) también pueden desempeñar un papel en la patogénesis a través de un proceso de mimetismo molecular. Modelos in vitro y en animales, que utilizan diferentes enfoques, apoyan el concepto de queuna interacción de PR3-ANCA con PR3 liberado de grânulos azurófilos y expresados en la superficie celular de neutrófilos TNF-a- activados, resultan enla desgranulación prematura de los neutrófilos, con el subsecuente daño endotelial y el reclutamiento de leucocitos2.
De otra parte, las infecciones (en especial por Staphylococcus aureus), el medio ambiente, y algunos químicos, tóxicos o fármacos, han sido sugeridos como desencadenantes de la enfermedad enpersonas con predisposición genética7.
Manifestaciones clínicas
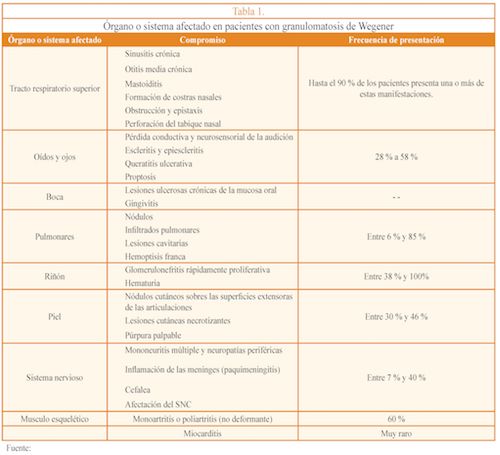
El espectro de presentación tiende a ser muy heterogéneo, siendo más común la presentación en oídos, nariz y garganta8. Las manifestaciones clínicas varían a menudo con el estadio de la enfermedad yel grado de afectación del órgano9. En la Tabla 1 se presentan los sistemas comprometidos, así como la afectación y frecuencia de presentación.
La enfermedad renal en ocasiones es la manifestación inicial, o se presenta durante el curso de la enfermedad. Puede progresar desde formas asintomáticas y leves a glomerulonefritis fulminante en cuestión de días o semanas, lo que resulta en insuficiencia renal terminal; incluso, con la terapia adecuada, la afectación puede conducir a insuficienciarenal crónicalO. De allí que hasta un 30% de lospacientes con enfermedad renal moderada a severa al momento del diagnóstico necesitarán terapia de reemplazo renal. Entre el 40 % y el 70 % de los pacientes puede recuperar la función renal, luego del tratamiento de inducción11.
Para medir la actividad de la enfermedad se ha planteado la utilización de herramientas como el score de Birmingham Vasculitis Activity Score for Granulomatosis with Polyangiitis (BVAS/GPA),que categoriza el compromiso de órganos y la actividad de la enfermedad en cada sistema, definiendoactividad como las anormalidades provocadas por la enfermedad de reciente diagnóstico o por el empeoramiento en las últimas cuatro semanas. Evalúa la actividad de la enfermedad en un punto de tiempo determinado como la suma de las manifestaciones del sistema de órganos individuales, definido poruna lista de 34 elementos ponderados causados por la enfermedad activa con respecto a si los síntomas son nuevos, peores o persistentes. Para la evaluación, cada uno de los 34 artículos se clasifica comosea, mayor o menor, dependiendo de la amenazainmediata para la vida o el órgano, con una calificación de 3 a 1 y una puntuación máxima combinada de 68. Los valores en ascenso de este score son un predictor negativo de supervivencia.
Cuando se evalúa la actividad de la enfermedad, se debe considerar lo inherente a la enfermedad activa y lo relacionado con el daño permanente ocasionado por la enfermedad. Las anormalidades producidas por la vasculitis que persisten por más de cuatro semanas se denominan enfermedad persistente y dan origen al daño que se define como los cambios irreversibles resultantes de la cicatrización y presentes por lo menos durante tres meses.
Diagnóstico
El diagnóstico se basa en los síntomas clínicos sugestivos. El patrón de oro es el estudio histológico en donde se observa vasculitis necrotizante de pequeños vasos e inflamación granulomatosa con células gigantes multinucleadas.
La identificación histopatológica rara vez se hace en las etapas tempranas de la GW. En pacientes consíntomas clínicos en los que la biopsia no es posible o no diagnóstica, la evaluación de ANCA positivo ayuda en el diagnóstico de estos casos. Los ANCA son anticuerpos cuyos objetivos son los dos componentes principales de los granulocitos neutrófilos: PR3 serina y mieloperoxidasa. Los anticuerpos anti-PR3 serina son prácticamente patognomónicos de la GW, mientras que los anticuerpos antimie-loperoxidasa son más sugestivos de otras vasculitis necrotizante primaria, principalmente poliangeítis microscópica2.
Hay dos tipos de pruebas para detectar ANCA: inmunofluorescencia y ensayo de inmunoabsorción ligado a enzimas (ELISA, por sus siglas en inglés).La inmunofluorescencia distingue entre anti-PR3y anti-mieloperoxidasa sobre la base de su patrónde tinción: la primera se asocia con c-ANCA yla segunda con p-ANCA. La estimación de los títulos de ANCA en el suero usando técnicas de inmunofluorescencia proporciona un diagnóstico confiable, a menudo sin necesidad de biopsia positiva. La sensibilidad de los títulos de c-ANCA depende de la actividad de la enfermedad2. El 20 % de los pacientes con GW activo presentan ANCA negativo. Este porcentaje aumenta hasta el 30 % enlas formas localizadas de la enfermedad2.
Tratamiento
Las terapias actuales minimizan la inflamación local y sistémica y pueden preservar la función del órgano. Los agentes inmunosupresores se combinan con manejo de soporte, que incluye: la compensación de la disfunción orgánica (tratamiento de la hipertensión o de diálisis); tratar o prevenir la comorbilidad (infección, osteoporosis o cataratas); empeoramiento de comorbilidad preexistente (empeoramiento de enfermedad cardíaca isquémica ola obesidad), o el desarrollo de nuevas patologías. El abordaje del paciente debe incluir la evaluación de la severidad y el contexto en que se produce la enfermedad. En la Tabla 2 se presentan los tratamientos inmunosupresores utilizados.
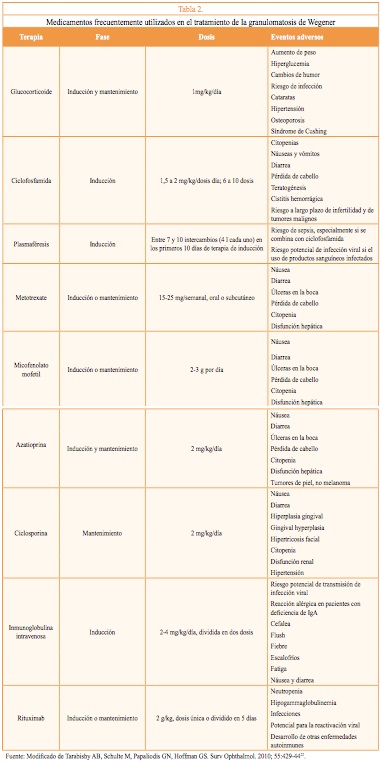
En general, los pacientes con GW responden bien a la terapia inmunosupresora con ciclofosfamida o metotrexato, asociado con glucocorticoides12. La dosis recomendada actual de ciclofosfamida es de 1,5 a 2 mg/kg/día y de 1 mg/kg/día de prednisona oral13.
Los pacientes reciben tratamiento de inducción intensivo seguido de terapia de mantenimiento. La mayoría de los pacientes requerirán terapia adicional para controlar comorbilidad y limitar la toxicidad del fármaco. Los tratamientos de inducción se pueden repetir para la recaída; sin embargo, puede ser necesario cambiar el tipo de inducción debido a la toxicidad o la pobre respuesta inicial.
Durante la etapa de remisión de la enfermedad, a lo largo del primer mes, la prednisona se titula hasta 5 mg/semana. Este régimen debe seguirse por un año después de la remisión completa, para luego reducirse gradualmente hasta suspender. La remisión completa puede tardar desde algunos meses hasta 1 o 2 años, con un tiempo medio de 12 meses. Con el uso del esquema de ciclofosfamida y corticosteroides se observa mejoría significativa en más del 90 % de los pacientes, logrando la remisión de la enfermedad en el 75 % de los casos. No obstante,se han observado recaídas hasta en el 50 % de los pacientes en remisión13.
El metotrexate y la azatioprina han sido utilizados en la fase de recuperación con buena respuesta; sin embargo, el metotrexate no debe utilizarse en pacientes con tasa de filtración glomerular de 50 ml/min o menor. También se han utilizado confrecuencia bolos de glucocorticoides para controlar reactivaciones. El micofenolato, la deoxispergualina y la leflunomida son otros fármacos que han demostrado ser prometedores en el mantenimientode la remisión.
El Rituximab ha sido recientemente utilizado durante la inducción y remisión. En el 2014 el National Institute for Health and Care Excellence (NICE) recomienda la utilización de Rituximab con glucocorticoide en los siguientes casos: si el tratamiento conciclofosfamida excede la dosis acumulada máxima, si está contraindicada o no es bien tolerada; si se prevé una posible alteración de la fertilidad; si la enfermedad se ha mantenido activa o ha progresado pese de un curso de 3 a 6 meses de tratamiento; si la persona ha tenido neoplasia urotelial14. Estas recomendaciones fueron tomadas a partir dos estudios: RAVE y RITUXVAS15,16. En el estudio RITUXVAS se reclutaron pacientes con afectación renal, quienes fueron aleatoriamente asignados a rituximab más ciclofosfamida (n = 33) o ciclofosfamida (n= 11). Ambos grupos recibieron metilprednisolonapor vía intravenosa (1 g) y un glucocorticoide oral (1 mg/kg/día inicialmente, reduciendo a 5 mg/día en el final de los 6 meses).
Los pacientes en el grupo rituximab recibieron infusiones de 375 mg/m2 por semana, durante 4 semanas, y ciclofosfamida intravenosa de 15 mg/kg, con la primera y tercera infusión de rituximab. Los pacientes en el grupo rituximab no recibieron tratamiento de mantenimiento. Los pacientes en el grupo de control recibieron ciclofosfamida intravenosa (15 mg/kg durante 3 a 6 meses, 6 a 10 dosis en total), seguido de azatioprina (2 mg/kg/día) como mantenimiento. También se les permitió el tratamiento con rituximab o ciclofosfamida, si los pacientes recaían.
La remisión sostenida se produjo en el 76 % de los pacientes en el grupo de rituximab más ciclofosfamida y en el 82 % de los pacientes del grupo de ciclofosfamida. La diferencia absoluta en remisión sostenida con rituximab más ciclofosfamida, en comparación con ciclofosfamida, fue de -6% (IC del 95 % -33 a 21). Entre los pacientes que todavía estaban en el estudio a los 12 meses, el 93 % de los del grupo de rituximab más ciclofosfamida y el 90% de los pacientes del grupo de ciclofosfamida estaban en remisión sostenida. No se encontró beneficio del rituximab sobre otros inmunomoduladores.
Otro ensayo clínico aleatorio comparó el tratamiento durante la fase de remisión del rituximab (n=57) y la azatioprina (n=58), luego de recibir manejo con ciclofosfamida y glucocorticoide. Los grupos recibieron 500 mg de rituximab en los días 0 y 14 y en los meses 6, 12 y 18 después del ingreso al estudio o azatioprina todos los días hasta el mes 22. Los autores demostraron la superioridad del rituximab (29 % frente a 3 % p=0,000), con frecuencia similar de eventos adversos en ambos grupos (n=25) (17).
La Guía de práctica clínica: vasculitis asociada a ANCA con compromiso renal realiza recomendaciones acerca del tratamiento de en GW sistémicas severas18, basándose en el tratamiento combinado: pulsos de metilprednisolona y ciclofosfamida como el tratamiento de primera línea para alcanzar la remisión. Asimismo, no recomienda el uso de ciclofosfamida a largo plazo, dado el mayor riesgo de recaídas; también señala que se debe tener en cuenta el recuento de glóbulos blancos pre y posinfusión.La recomendación del uso de rituximab es la misma que la presentada en las guías NICE. El uso de metotrexate y glucocorticoide puede darse como alternativa a la ciclofosfamida en diagnóstico temprano sistémico y FG > 60ml/min/1,73 m2 de superficie corporal, pero es menos eficaz en el control de la enfermedad (recaídas) durante el mantenimiento. En insuficiencia renal y/o hepática el uso de metotrexate está contraindicado, siendo en estos casos el micofenolato más glucocorticoide una opción terapéutica alternativa. La plasmaféresis, como terapiaco adyuvante, estaría indicada en casos severos conglomerulonefritis rápidamente proliferativa, a fin de mejorar la supervivencia renal o requerimiento dialítico y/o hemorragia pulmonar.
Las inmunoglobulinas intravenosas (IglV) han demostrado eficacia terapéutica potente en pacientes con vasculitis ANCA positiva; son consideradas como una alternativa terapéutica en pacientes con enfermedad refractaria o en pacientes en los que la terapia convencional está contraindicada, por ejemplo, cuando hay presencia de infección, en pacientes gravemente enfermos o durante el embarazo. En el momento no se encuentran guías acerca de la duración, frecuencia o dosis óptima de IglV; algunos autores utilizan este régimen con base en pequeñas series publicadas, asociándolo a corticoides, hastaque los agentes inmunosupresores ya no se encuentren contraindicados8.
Una revisión Cochrane identificó un ensayo clínico aleatorio (ECA) al que concurrieron 34 participantes que fueron asignados para recibir IglV (un ciclo único de 400 mg/kg/día durante 5 días) o placebo, además de azatioprina y corticoide sistémico para el mantenimiento de la remisión19. Se encontró que no hubo diferencias significativas al comparar IgIV adyuvante con placebo adyuvante en los desenlaces mortalidad, en eventos adversos graves, en el tiempo hasta la recaída y en la infección. La disminución en la severidad de la enfermedad derivada de los síntomas informados por los pacientes, fue ligeramente mayor en el grupo de IgIV que en elgrupo placebo, al mes y a los tres meses. También hubo un aumento significativo en el total de eventos adversos en el grupo de IgIV (riesgo relativo: 3,50; IC del 95 %: 1,44 a 8,48; p <0,01). Los autores concluyeron que el ECA no suministra evidencia suficiente que permita asegurar que la terapia adyuvante con IgIV proporciona una ventaja terapéutica, comparada con la combinación de esteroides e inmunosupresores para pacientes con GW.
La plasmaféresis ha sido reconocida como un tratamiento de segunda línea. De acuerdo con la Sociedad Americana de Aféresis (ASFA, por sus siglas en inglés), la plasmaféresis es una indicaciónde clase I para GW en los casos dependientes de diálisis y categoría III en los casos no dependientesde diálisis20. Malhotra, et al21 reportaron el caso de un paciente no dependiente de diálisis en el que se realiza tratamiento conjunto de plasmaféresis con rituximab, y se logra alcanzar la remisión luego de no responder a la terapia con altas dosis de ciclofosfamida. En este paciente, al no presentar mejoría con ciclofosfamida, se decidió iniciar rituximab. Sin embargo, como el pico de acción de rituximab se produce a las 3-4 semanas después de la administración, se inició plasmaféresis para manejo del período agudo y crítico.
La Velocidad de sedimentación globular (VSG) y los niveles de c-ANCA se utilizan para vigilar la actividad de la enfermedad y el diagnóstico precoz de la recidiva; sin embargo, existe discusión acercade su verdadera utilidad.
La GW puede asociarse a una mortalidad temprana, especialmente en pacientes con compromiso renal que no reciben tratamiento inmunomodulador temprano. Varias modalidades de tratamiento se han empleado dependiendo de los órganos y sistemas comprometidos. Aunque se recomienda como terapia inicial el uso de ciclofosfamida más esteroide sistémico, los eventos adversos y la alta tasa de recaída no dejan de manifiesto que sea la mejor terapia para uso sistemático; es más, no están bien definidas las dosis y duración de las mismas.
En nuestro paciente se observó una clásica presentación de compromiso crónico de las vías aéreas, la afectación pulmonar bilateral y la insuficiencia renal progresiva. Los hallazgos clínicos y paraclínicos arrojaron un compromiso infeccioso sistémico, que pudo agravar el compromiso renal. El escalonamiento antibiótico empírico fue el adecuado, de acuerdo a las recomendaciones institucionales; sin embargo, la sospecha de enfermedad autoinmune formulada por el servicio de nefrología tienen un impacto importante y significativo en la evolución de la paciente. Aunque el uso de ciclofosfamida se considera el tratamiento de primera línea, es importante valorar el contexto de cada paciente (individualizar). En nuestro caso se decidió el uso de rituximab para evitar el compromiso de la inmunidad celular, dado que la paciente manifestó neutropenia.
El mantenimiento óptimo de la remisión en Granulomatosis con poliangeitis (GPA) y otras Vasculitis asociadas a ANCA (AAV) sigue siendo un tema de debate. Debido al compromiso renal, consideramos que el tratamiento con rituximab en nuestro paciente es una excelente elección que se encuentra acorde con lo revisado en la literatura, representado menor riesgo de recaída y de complicaciones secundarias.
El tratamiento de la vasculitis ha mejorado, pero hasta que se pueda controlar por completo o curar la enfermedad, sigue siendo insatisfactorio. Es posible prevenir la mortalidad temprana y se han reducido los efectos inmediatos de la vasculitis activa en la función del órgano. Sin embargo, nuestro objetivo es ampliar aún más la probabilidad de supervivencia, así como mejorar la calidad de vida de los pacientes que sobreviven a las presentaciones severas, asegurando que se minimiza la actividad de la enfermedad y el daño, la toxicidad del fármaco y el deterioro de la calidad de vida.
Conclusiones
El compromiso renal en pacientes con GW es común y puede llegar a ser letal. Una alta proporción de los pacientes puede llegar a requerir terapia de reemplazo renal, aunque gran parte logra retornar a una función renal normal. No están claros los factores que se asocian al retorno de función renal normal, pero sí se ha descrito que depende de manejos tempranos y a altas dosis de inmunomoduladores.
Ahora, con relación a los esquemas de tratamiento, se han basado en ensayos clínicos con un pequeño número de participantes y enestudios observacionales con riesgo de sesgo, por lo que el tratamiento ideal (menor tasa de recaída, menores efectos adversos, menor mortalidad, mayor curación) todavía no está completamente definido. El rituximab es un medicamento que se está utilizando a mayor escala que otros tratamientos convencionales, con efectividad adecuada y menores efectos secundarios en estos pacientes. Cabe señalar que dos guías de práctica clínica tienen recomendaciones claras acerca de sus usos, aunque tales recomendaciones no aplicana todos los pacientes, en especial para el caso manejado en nuestra institución. Es por ello que consideramos que se requieren más estudios que evalúen el tratamiento con rituximab en pacientescon compromiso severo y otras comorbilidades.
Responsabilidades éticas
Protección de personas y animales
Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos
Los autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informado
Los autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de intereses
Declaramos en el presente manuscrito no tener conflictos de interés y que la publicación del mismo se hace con fin de aportar al conocimiento de la comunidad médica y científica.
Referencias
1. Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclature of systemicvasculitis. Proposal of an international consensus conference. Arthritis Rheum. 1994 Feb;37(2):l ST-92.
2. Iannella G, Greco A, Granata G, Manno A, Pasquariello B, Angeletti D, et al. Granulomatosis withpolyangiitis and facial palsy: Literature review and insight in the autoimmune pathogenesis. Autoim-mun Rev. 2016 Jul; 15(7):621-31.
3. Liu X, Cui Y, Li Y, Wang C, Zhao H, Flan J. Using inpatient data to estimate the prevalence of Wegener’s granulomatosis in China. Intractable Rare Dis Res. 2016 Feb;5(l):31-5.
4. Wallace ZS, Lu N, Unizony S, Stone JH, Choi F1K. Improved survival in granulomatosis with polyangiitis: A general population-based study. Semin Arthritis Rheum. 2016 Feb;45(4):483-9.
5. Romeu M, Couchoud C, Delarozière JC, Burtey S, Chiche L, Harlé JR, et al. Survival of patients withANCA-associated vasculitis on chronic dialysis: data from the French REIN registry from 2002 to 2011. QJM. 2014 Jul;107(7):545-55.
6. Hollé JU, Gross WL. Treatment of ANCA-associated vasculitis (AAV). Autoimmun Rev. 2013Feb;12(4):483-6.
7. Kronbichler A, Kerschbaum J, Mayer G. The Influence and Role of Microbial Factors in Autoimmune Kidney Diseases: A Systematic Review. J Immunol Res. 2015;ID:858027.
8. Guidelli GM, Tenti S, Pascarelli NA, Galeazzi M, Fioravanti A. Granulomatosis with polyangiitis and intravenous immunoglobulins: a case series and review of the literature. Autoimmun Rev. 2015Aug;14(8):659-64.
9. Wojciechowska J, Krajewski W, Krajewski P, Krçcicki T. Granulomatosis With Polyangiitis in Otolaryngologist Practice: A Review of Current Knowledge. Clin Exp Otorhinolaryngol. 2016 Mar;9(l):8-13.
10. Shafiei K, Luther E, Archie M, Gulick J, Fowler MR. Wegener granulomatosis: case report and briefliterature review. J Am Board Fam Pract. 2003 Nov-Dec;16(6):555-9.
11. Lee T, Gasim A, Derebail VK, Chung Y, McGregor JG, Lionaki S, et al. Predictors of treatmentoutcomes in ANCA-associated vasculitis with severe kidney failure. Clin J Am Soc Nephrol. 2014May;9(5):905-13.
12. Cavoli GL, Ferrantelli A, Bono L, Tortorici C, Passantino R, Rotolo U. Kidney involvement in a Wegener granulomatosis case. Indian J. Med Sci. 2012 Sep-Oct;66(9-10):238-40.
13. Sada K-e, Yamamura M, Harigai M, Fujii T, Takasaki Y, Amano K, et al. Different responses to treatment across classified diseases and severities in Japanese patients with microscopic polyangiitis and granulomatosis with polyangiitis: a nation wide prospective inception cohort study. Arthritis Res Ther.2015;17(1):1-12.
14. Latimer N, Carroll C, Wong R, Tappenden P, Venning M, Luqmani R. Rituximab in Combination with Corticosteroids for the Treatment of Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis: ANICE Single Technology Appraisal. Pharmacoeconomics. 2014 Dec;32(12):1171-83.
15. Jones RB, Tervaert JW, Hauser T, Luqmani R, Morgan MD, Peh CA, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010 Jul;363(3):211-20.
16. Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010; Jul;363(3):221-32.
17. Guillevin L, Pagnoux C, Karras A, Khouatra C, Aumaitre O, Cohen P, et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med. 2014 Nov;371(19):1771-80.
18. Vázquez V, Fayad A, González G, Quevedo AS, Sindin JR. Vasculitis asociada a ANCA con compromiso renal. Guía de Práctica Clínica. Medicina (Buenos Aires). 2015;75(Suplemento I):1-38.
19. Fortin PM, Tejani AM, Bassett K, Musini VM. Intravenous immunoglobulin as adjuvant therapy for Wegener’s granulomatosis. Cochrane Database Syst Rev. 2013 Jul;(3): CD007057.
20. Schwartz J, Winters JL, Padmanabhan A, Balogun RA, Delaney M, Linenberger ML y col. Guidelines on the use of therapeutic apheresis in clinical practice—evidence-based approach from the writing committee of the american society for apheresis: the sixth special issue. J Clin Apher. 2013Jul;28(3):145-284.
21. Malhotra S, Dhawan HK, Sharma RR, Marwaha N, Sharma A. Successful Management of Refractory Dialysis Independent Wegener’s Granulomatosis with Combination of Therapeutic Plasma Exchangeand Rituximab. Indian J Hematol Blood Transfus. 2016 Jun;32(Suppl 1):332-4.
22. Tarabishy AB, Schulte M, Papaliodis GN, Hoffman GS. Wegener’s granulomatosis: Clinical manifestations, differential diagnosis, and management of ocular and systemic disease. Surv Ophthalmol.2010; 55:429-44.